
 CPSL Partners
CPSL Partners Clients
Clients
 Français
Français Deutsch
Deutsch Español
Español English US
English USInternational Translation and Localisation Blog
Accurate communication tailored to each audience.
Make the CPSL Life Sciences team part of your Master Implementation Plan for the new Medical Devices Regulation
Building a roadmap for implementation of the new EU Medical Devices Regulation means defining sub-projects like translations, resource requirements, and a steering group, while ensuring overall responsibility. This major undertaking has been hindered by the COVID-19 pandemic, so much so that the European Commission has issued the “Proposal for a Regulation of the European Parliament and of the Council amending Regulation (EU) 2017/746 as regards transitional provisions for certain in vitro diagnostic medical devices and deferred application of requirements for in-house devices” to give the industry a little more time to adapt.
The new Medical Devices Regulation encompasses a wide range of laboratory developed tests, point-of-care devices, instruments, reagents and kits used to analyse human samples and guide clinical decision-making and is far more rigorous and detailed. All these have a crucial role in saving lives by providing innovative medical solutions for the diagnosis, prevention, monitoring, prediction, prognosis, treatment and alleviation of disease. It is estimated 70% of clinical decisions are made using in vitro diagnostic medical devices.
Because this is such an exciting, fast-moving field, the European Union has reversed its previously lenient stance by introducing the Diagnostic Medical Device Regulation. It is a robust, transparent, predictable and sustainable regulatory process envisaged to raise quality and safety standards with clearer definitions and scope, improving oversight and traceability across the supply chain throughout the device lifecycle. All this will enable the industry to accommodate recent technologies and scientific advances and facilitate global trade by promoting regulatory convergence.
The pandemic, notification and NANDO
The new regulation calls for reinforcement of the criteria for the designation and overseeing of notified bodies (a “notified body” is an independent, third-party conformity assessment body). Under the NANDO (New Approach Notified and Designated Organisations) Information System, notification is an act whereby a Member State informs the Commission and the other Member States that a body, which fulfils the relevant requirements, has been designated to carry out conformity assessment according to a directive. Notification of Notified Bodies and their withdrawal are the responsibility of the notifying Member State.
The best laid plans of mice and men often go awry (Robert Burns, 1785), and the pandemic unexpectedly diverted resources and caused delays. There were initially only six notified bodies designated to date under the In Vitro Diagnostic Medical Devices Regulation. This critical shortage of notified body capacity made it difficult for manufacturers to complete the requisite conformity assessment procedures.
Breathing space
Despite considerable progress in recent years, the overall capacity of conformity assessment bodies (notified bodies) remains insufficient for carrying out their tasks. In addition, many manufacturers are not sufficiently prepared to meet the strengthened requirements of the MDR, thereby threatening the availability of medical devices in the EU market.
There has been an increase in the number of formerly designated notified bodies, present in only three countries (Germany, France, and the Netherlands), given that, according to the “European Parliament resolution on the urgent need to revise the Medical Devices Regulation (2024/2849(RSP))”, dated 16 October 2024, the “notified body capacity for the MDR has now reached 50”, although “only 13 notified bodies have been designated by Member States to carry out assessments under the IVDR”. This is probably part of the reason why the deadlines have been extended even further.
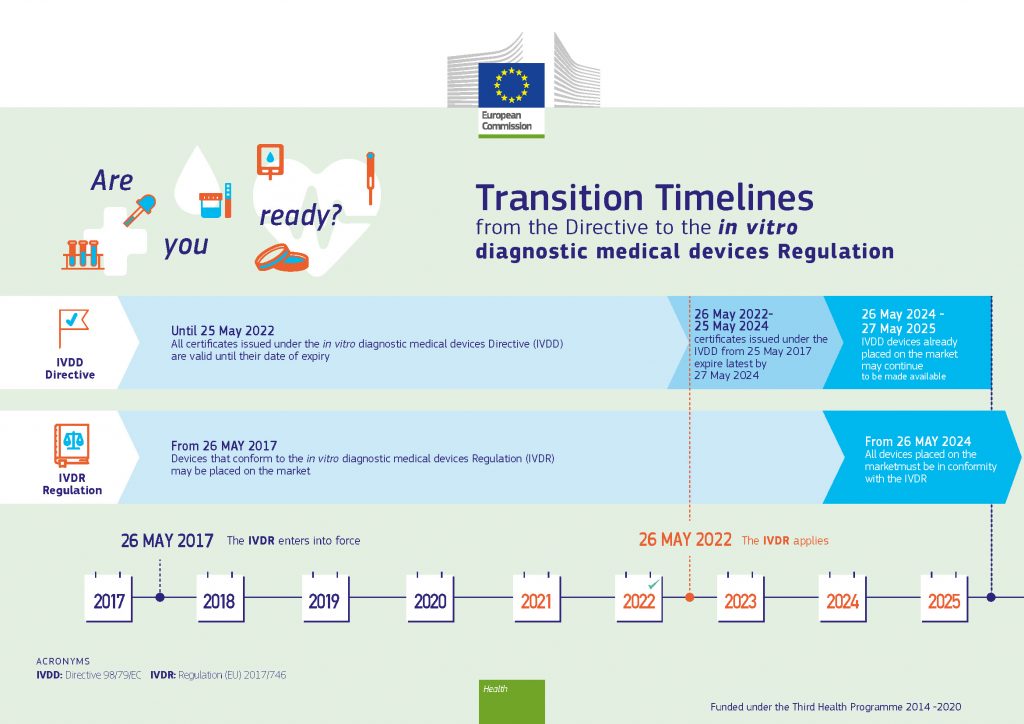
The MDR came into force on 26 May 2021, and the “transition period” ended on 26 May 2024. The IVDR has been applicable since 26 May 2022. However, in January 2022 the European Parliament and the Council adopted a staggered extension of the IVDR’s transition period, ranging from 26 May 2025 for higher risk in vitro diagnostics, to 26 May 2027 for lower risk in vitro diagnostics, and to 26 May 2028 for certain provisions concerning devices manufactured at and used in health institutions.
Further updates introduced by Regulation (EU) 2024/1860 also include the gradual rollout of the EUDAMED database and a new obligation for manufacturers to inform authorities in the event of interruption or discontinuation of the supply of devices. In addition, the reassessment interval for notified bodies has been extended to five years, thereby easing the administrative burden without reducing oversight.
Our experts are here to lighten the load
Despite the postponement, the countdown to implementation of the In Vitro Diagnostic Medical Devices Regulation continues, and the current directive, IVDD 98/79/EC phased out. That’s why CPSL has a dedicated Life Sciences team working to lighten the load. Industry players are finding CPSL’s expert services invaluable for successfully navigating the demands of the new regulatory standards. The Life Sciences team at CPSL is poised to smooth out the transition to all kinds of changes in this rapidly developing industry, and it is a trusted language partner for bearing the burden of managing time-consuming tasks, leaving your experts free to conquer new challenges with clarity and confidence.
The industry has been granted a little more breathing room to account for the effects of the pandemic. That said, the basic requirements have not changed. One feature of the new law is the introduction of a risk-based classification system with four risk classes of in vitro diagnostic medical devices: class A (low individual risk and low public health risk), class B (moderate individual risk and/or low public health risk), class C (high individual risk and/or moderate public health risk), and class D (high individual risk and high public health risk). However, the length of the new proposed transition periods now depends on the type of device, as per (EU) 2024/1860, of 13 June 2024:
- Devices with a certificate that was issued in accordance with Directive 98/79/EC and that is valid by virtue of paragraph 2 of this document may be placed on the market or put into service until 31 December 2027.
- Devices for which the conformity assessment procedure pursuant to Directive 98/79/EC did not require the involvement of a notified body, for which a declaration of conformity was drawn up prior to 26 May 2022 in accordance with that Directive, and for which the conformity assessment procedure pursuant to this Regulation requires the involvement of a notified body may be placed on the market or put into service until the following dates:
31 December 2027 for class D devices;
31 December 2028 for class C devices;
31 December 2029 for class B and class A devices placed on the market in sterile condition.
CPSL can provide the transition assistance you need
Linguistic excellence is always non-negotiable, but especially in medical and regulatory matters in which everything must be precisely drafted and verified to provide the highest of guarantees in matters of life and death. The new regulation accounts for this. For example:
Article 10(10), on the General obligations of manufacturers, states that “Manufacturers shall ensure that the device is accompanied by the information […..] in an official Union language(s) determined by the Member State in which the device is made available to the user or patient. The particulars on the label shall be indelible, easily legible and clearly comprehensible to the intended user or patient. The information supplied in accordance with Section 20 of Annex I with devices for self-testing or near-patient testing shall be easily understandable and provided in the official Union language(s) determined by the Member State in which the device is made available to the user or patient.”
Article 37 of the new regulation continues to set out language requirements and states that “All documents required pursuant to Articles 34 and 35 shall be drawn up in a language or languages which shall be determined by the Member State concerned. Member States, in applying the first paragraph, shall consider accepting and using a commonly understood language in the medical field, for all or part of the documentation concerned. The Commission shall provide translations of the documentation [……], or parts thereof into an official Union language, such as is necessary for that documentation to be readily understood by the joint assessment team appointed in accordance with Article 35(3).”
The third revision of the language requirements for MD manufacturers (August 2025) summarises the national provisions. Other language requirements are stated in MDCG 2019-9 – Rev.1 – Summary of safety and clinical performance Guidance Document. Accordingmanufacturers for implantable and class III medical devices. SSCPs must be written in a way that is clear to the intended user and, if relevant, to the patient, and they must be translated into the languages accepted in the Member States where the device is intended to be sold (page 6). SSCPs are publicly available documents stored in the EUDAMED.
The European Union has published a series of dedicated factsheets that summarise the principal areas of activities in the medical devices sector for manufacturers of medical devices and in vitro diagnostic medical devices, and the EU is urging the industry to act promptly due to the expected pressure on notified bodies. As a global leader in serving the Life Sciences sector, we are fully committed to streamlining regulatory compliance by meeting all your needs – translation, revision, adaptation, and back-translation (We master the latest technologies to ensure consistency and effective terminology management, while following strict translation protocols for guaranteed excellence.
If you need assistance with this crucial process, CPSL is your language provider of choice: download our guide to the new MDR now or get in touch to find out how we can assist you.
CPSL has recruited and appointed an expert team to undertake these highly technical, crucially important tasks for Life Sciences companies, all coordinated by dedicated project managers with extensive industry experience.


Your gateway to Europe...Don't wait until the last minute!

Get your MD and IVD EU Regulations just right!
Download your Whitepaper now
E-learning localisation

Let’s talk about your company’s next online course today
What are you waiting for?







